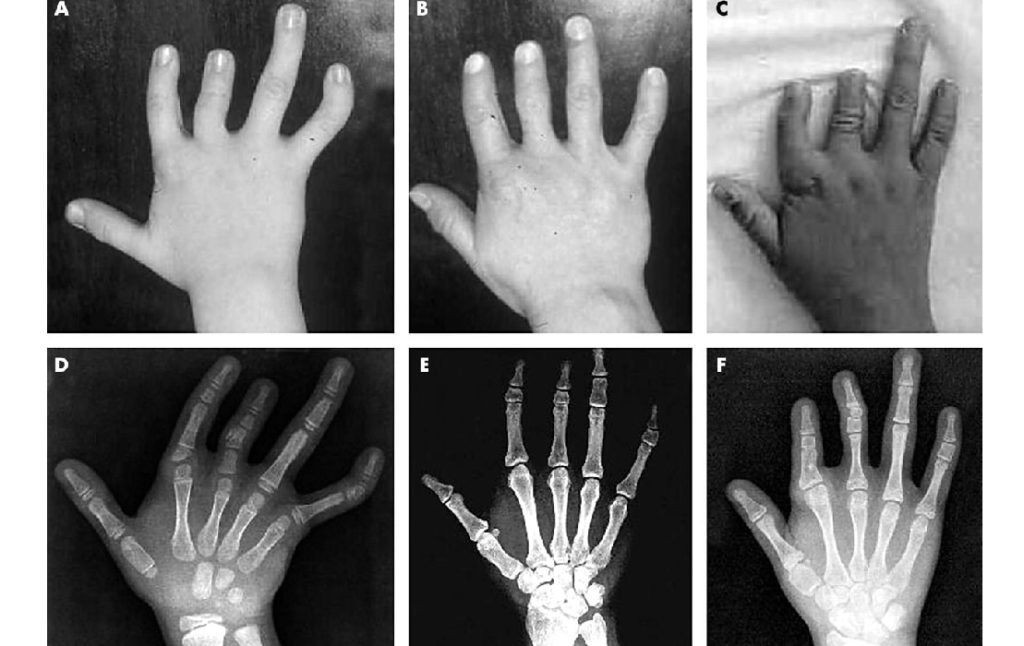
Лечение синдактилии
Лечение синдактилии возможно исключительно хирургическим путём. Из-за разнообразия форм, типов и видов заболевания оптимальную методику подбирают строго индивидуально, учитывая его особенности в каждом отдельном случае. Оперативное вмешательство не только направлено на устранение косметического эффекта, но и позволяет улучшить функции поражённой конечности. При сложных формах оно требует проведения нескольких пластических операций, корректирующих кожный покров, мягкие ткани и кость.
Разрабатывая план операции, хирург принимает во внимание тот факт, что разъединение пальцев должно осуществляться по этапам. Так, если имеет место синдактилия всех или нескольких пальцев, прежде всего отделяют первый и пятый, потом — второй и третий и только после этого — третий и четвёртый
Такой подход оправдан, поскольку исключает риск отмирания в случае если во время инвазии будет повреждён крупный кровеносный сосуд.
Ещё один принцип, которым руководствуются хирурги, — исключение натяжения раневых краёв, кожа должна быть сопоставлена свободно. В противном случае сформируются грубые, некрасивые рубцы и перетяжки, причём последние не позволят пальцам развиваться нормально и приведут к появлению контрактур.
Несмотря на то, что чёткой систематизации операций при синдактилии не существует из-за разнообразия дефектов, можно выделить пять их основных групп. Они представлены рассечением:
- тонкой связки между пальцами без коррекции кожного покрова;
- кожной перепонки с пластикой местными тканями;
- сросшихся между собой пальцев с пластикой кожи расщеплённым лоскутом;
- сросшихся между собой пальцев с мероприятиями кожной ластики с применением местных тканей и тканевых трансплататов, взятых с другого участка тела;
- со многими этапами и проведением кожной, мышечной, .
Через полмесяца после вмешательства начинают восстановительное лечение с применением консервативных методик: массажа кистей/стоп, лечебной физкультуры, электростимуляции, аппликаций.
Хотите быть уверены, что хирурги сделают всё зависящее от них и приложат все усилия к тому, чтобы добиться наилучших результатов при синдактилии? Обращайтесь в ЦЭЛТ!
Диагностика
Установить точный диагноз врачу помогают лабораторные исследования, которые проводятся, когда малыш находится еще в утробе матери. Дополнительные тесты назначает врач акушер гинеколог. Когда малыш появился на свет, диагностикой и лечение занимается ортопед.
| Название | Описание |
| Первичный осмотр |
|
| Инструментальные методы диагностики |
|
| Генетические тесты |
|
Максимально информативный и окончательный анализ, который помогает установить правильный и точный диагноз – это тест ДНК. Будущая мама должна знать, что брахидактилия не является причиной, чтобы прервать беременность. Ситуация усугубляется, если вместе с короткопалостью обнаружены сопутствующие генетические заболевания.
Особенности препарирования при минимальном инвазивном вмешательстве
У препарирования при минимальных инвазивных вмешательств есть ряд особенностей:
- Достаточное водяное охлаждение в объеме не меньше 70 мл/мин. Идеальным вариантом будет турбинный наконечник, оснащенный 3-4 отверстиями для подачи спрея;
- Идеальный контроль твердых тканей зуба на протяжении всего препарирования. Обычно для этого используются очки-линзы или увеличивающие стоматологические зеркала;
- Применение кариес-маркеров для контроля удаления всех тканей, пораженных кариесом.
После препарирования полости обычно имеют уникальный, произвольный дизайн, при котором сохранено максимальное количество здоровых твердых тканей. Смазанный слой, которые образуется в полости после препарирования и мешает адгезивному материалу связываться с тканями зуба, удаляется ортофосфорной кислотой, кондиционером или самопротравливанием композитов.
Виды
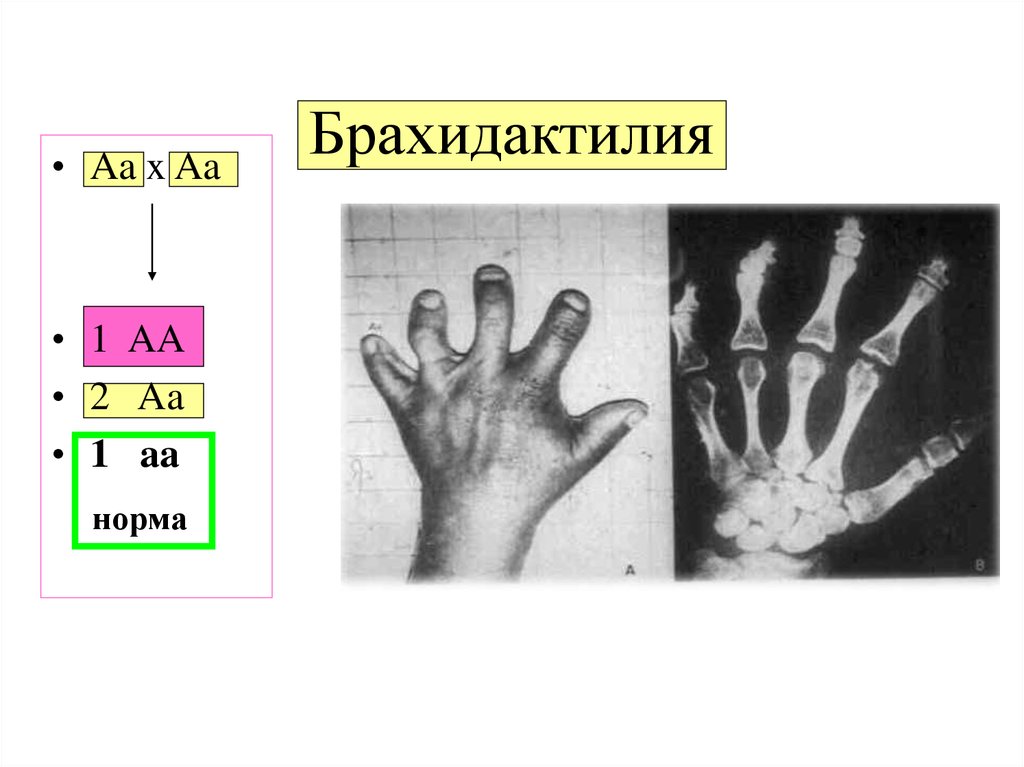
Разделения брахидактилии выражаются разными степенями развития и, поэтому, делятся на следующие типы:
1) Тип А, когда средние пальцы руки укороченные и искривленные, появляется дисплазия пластинок ногтей;
2) Тип В, когда фаланги на концах стоп и кистей являются недоразвитыми, видно как второй и третий палец сращиваются, есть патологии при образовании зубов, костей позвоночника и черепа;
3) Тип С, когда мы замечаем недоразвитие пястных костей, средних фаланг пальцев. У детей, страдающих данной патологией наблюдались низкий рост и торможение в умственном развитии;
4) Тип D, когда большие пальцы конечностей человеческого организма сокращаются в размерах;
5) Тип Е – при данном типе ключица и пястные кости не развиваются;
Сегодня одной из самых тяжелых форм заболевания брахидактилии считается тип B, самым редко встречающимся типом – Е, а встречающийся чаще всего случаи заболевания типа D.
Профилактика
В большинстве случаев патология не требует тщательной диагностики. Аномалия видна сразу после рождения, и определяется педиатром. Поскольку патология может привести к серьезным ограничениям и дискомфорту в повседневной жизни человека, к врачу следует всегда обращаться, если у ребенка развивается серьезная инвалидность. Таким образом можно избежать дополнительных осложнений и трудностей во взрослой жизни.
При возникновении серьезных психических дисфункций или комплекса неполноценности необходимо обратиться к врачу, чтобы избежать депрессии.

Депрессивный синдром
Существует один способ предотвратить эту деформацию в медицинском смысле. Недостаточное питание, никотин и алкоголь во время беременности и в целом нездоровый образ жизни может способствовать появлению пороков у ребенка. Наркотики и лекарства, отпускаемые по рецепту, усугубляют ситуацию.
Прямых методов по предотвращению болезни не существует
При появлении у ребенка дополнительных симптомов или сопутствующих патологий важно обращаться к доктору. Своевременное лечение аномалий развития может смягчить течение болезни во взрослой жизни
Неправильная терапия в раннем возрасте может усугубить течение болезни. При любых симптомах или аномалиях, возникших у ребенка, нужна консультация врача. Своевременное лечение помогает уменьшить симптоматику и подготовить суставы ребенка.


Похожее:
- Причины возникновения родовых травм у новорожденных в области шейного отдела позвоночника, последствия, симптоматика, методы диагностики и лечения
- Причины возникновения молоткообразной деформации, лечение второго пальца, методы диагностики, профилактики, симптомы и потенциальные осложнения
- Причины возникновения шишки на большом или маленьком пальце руки, симптоматика, диагностика, методы лечения, профилактики и потенциальные осложнения
- Причины возникновения боли в области лучезапястного сустава, сопутствующая симптоматика, методы диагностики, лечения и профилактики
- Причины возникновения артрогрипоза у маленьких детей, патогенез, основные симптомы, методы диагностики, лечение и профилактика
- Причины возникновения узелков Бушара и Гебердена, симптоматика, методы диагностики, консервативного лечения, оперативного вмешательства и прогноз
- Симптомы бурсита пальцев стопы, методы лечения, причины возникновения, патогенез, осложнения и прогноз болезни
Как выбирается пломбировочный материал
При минимальном инвазивном лечении кариозные полости, как правило, имеют сложную форму и малые размеры, кроме того, доступ к ним обычно затруден. В связи с этим материал, который должен заполнить такой дефект, должен быть достаточно и иметь низкий модуль упругости.
Всеми этими свойствами обладают текучие реставрационные материалы, которые делятся на несколько групп.
По химическому составу
- Жидкие композиты: Flow Line, Synergy Flow, Revolution, Filtek Flow, Flow It!;
- Жидкие компомеры: Prima Flow, Dyract Flow;
- Жидкие ормокеры: Definite Flow, Admira Flow,;
- Герметики.
По консистенции
- Низкотекучие: Gradia LoFlow, Filtek Supreme XT Flow, Alpha Flow, EsthetX Flow, Point4;
- Среднетекучие: X-flow, Filtek Flow, Gradia Flow, Flow It! LF;
- Сильнотекучие: Flow Line, Revolution, Flow It!.
По способу полимеризации
- Химического отвердения: Self Cure, Flow It!;
- Светового отвердения: Revolution, Filtek Flow, Flow It! и ряд других.
Показания к применению текучих пломбировочных материалов
- Ямки, фиссуры и трещин;
- Полости III и V классов;
- Маленькие полости первого класса с небольшой жевательной нагрузкой;
- Полости второго класса после бокового доступа или туннельного препарирования;
- Небольшие дефекты в непрямых и прямых реставрациях, которые требуют восстановления.
Работа с текучими материалами
Во время работы с текучими композитными материалами очень важно учитывать то, что они имеют достаточно высокий процент усадки – в общем около 5%. Также при неправильной технике пломбирования могут образоваться поры, а контролировать границы реставрации становится достаточно сложно
Для внесения композитных материалов следует применять капсулы с поршнем, вставляя их в обычный пистолет для капсульных пломбировочных материалов. С помощью такой техники можно герметично и без образования пор заполнить полость любой конфигурации даже с ограниченным доступом.
После пломбирования осуществляется окончательная пришлифовка пломбы и проверка окклюзионных контактов. Если реставрация осматривается повторно, то для ее оценки применяют ряд критериев:
- Общая сохранность реставрации;
- Краевое окрашивание;
- Краевое прилегание;
- Цветостабильность;
- Качество поверхности;
- Возникновение вторичного кариеса.
Очень важно постоянно контролировать реставрацию в мини-полости – это позволит сохранить ее на долгие годы, а при обнаружении проблем снова воспользоваться малоинвазивными методами лечения
Операция брахидактилии
Современная медицина предлагает больным с генетическими аномалиями исключительно оперативное лечение. Врач может полностью заменить или удлинить слаборазвитые фаланги на пальцах. После операции пациентам рекомендуется заниматься физиотерапевтическими процедурами. Гимнастика и упражнения помогут пациенту заново научиться управлять руками и ногами.

Хирургическое лечение больным показано при следующих обстоятельствах:
- серьезный внешний дискомфорт;
- нарушение функционирования рук и ног;
- сращивание пальцев;
- кисти и стопы короткие.
Для устранения дефектов и восстановления функций проводят следующие виды оперативного вмешательства:
| Название | Описание |
| Поллизация | Хирург перемещает 1 из пальцев на место большого. |
| Дистракция | Во время медицинских манипуляций врач растягивает костную ткань, которая не полностью развилась. |
| Аутотрансплантация | Осуществляется пересадка пальцев. |
| Пластика (синдактилия) | Операция позволяет разделить смежные элементы на костях, коже, сухожилиях или мышцах. |
Оперативное вмешательство проводится, если ребенку исполнилось 3 года. Метод хирургического лечения подбирает врач ортопед. Специалист учитывает степень развития патологических процессов и состояние костной системы детского организма.
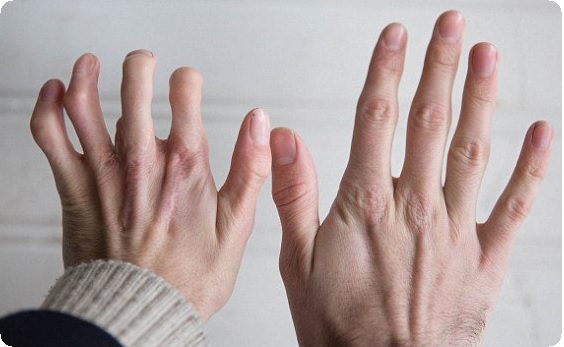
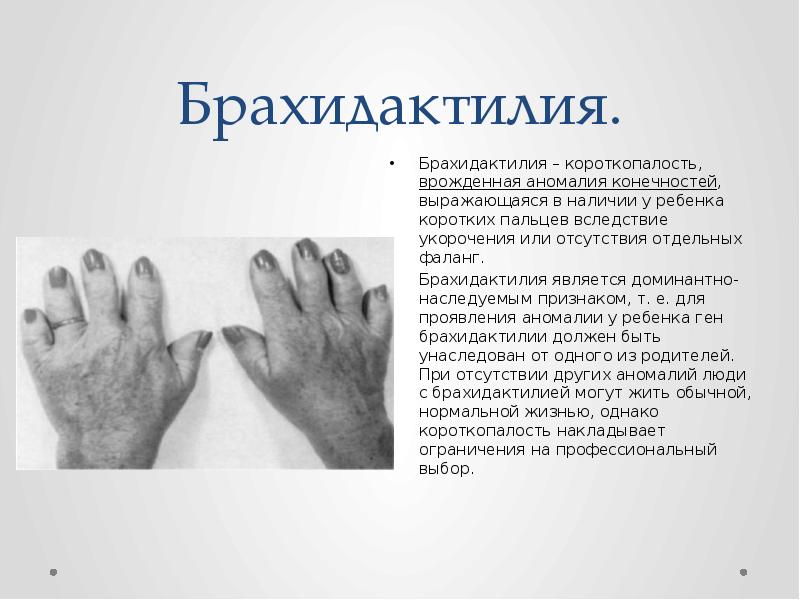
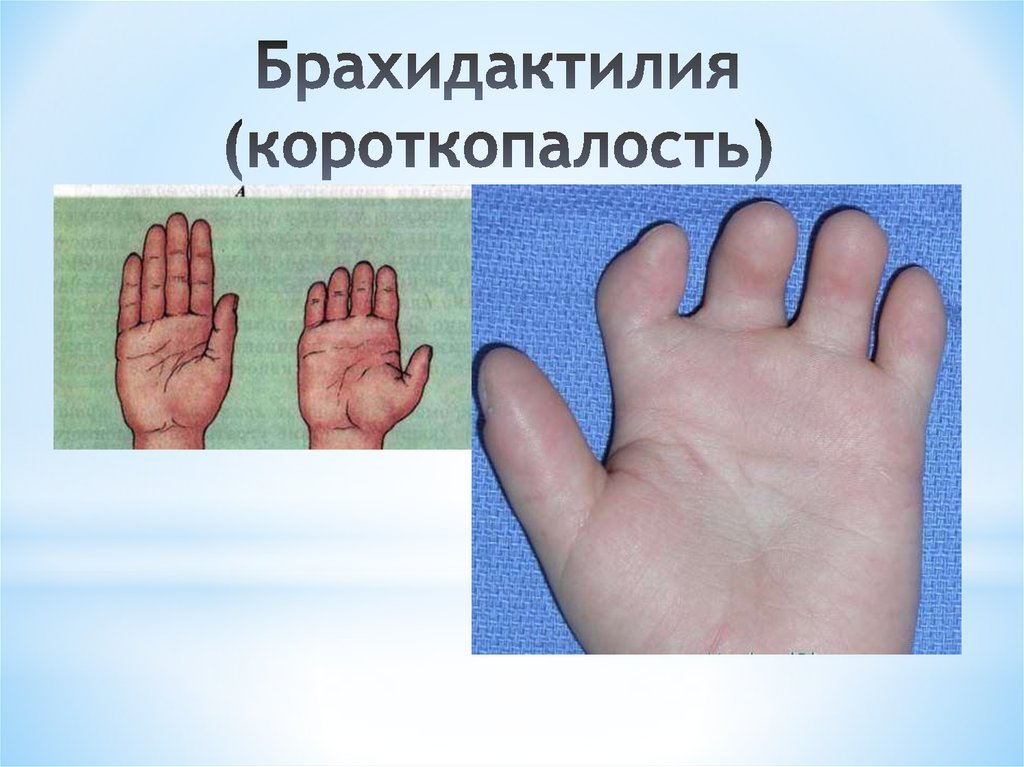


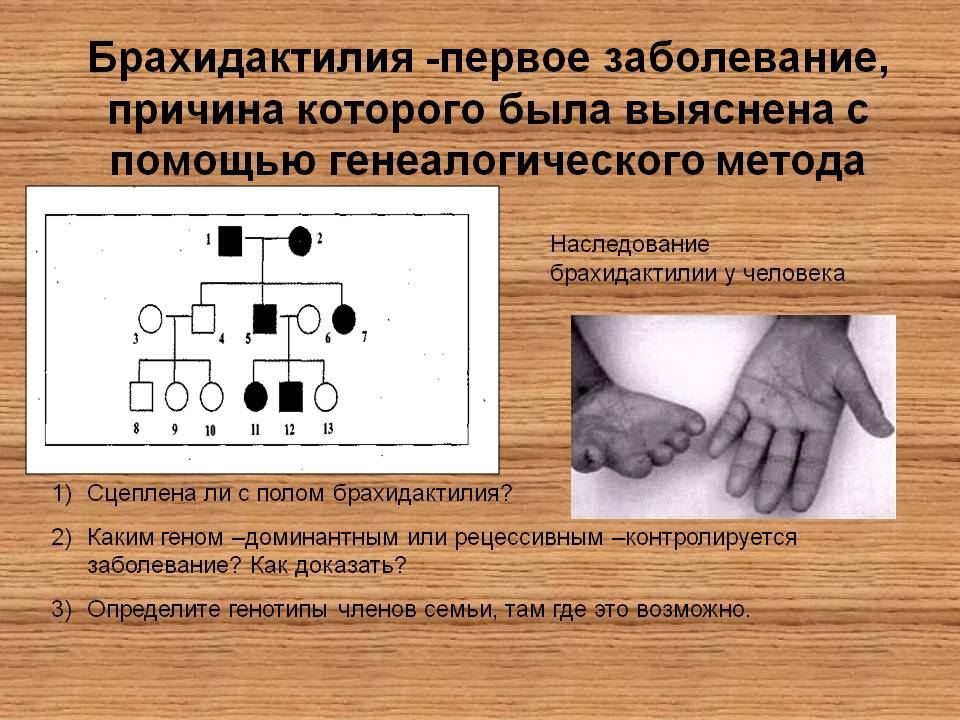
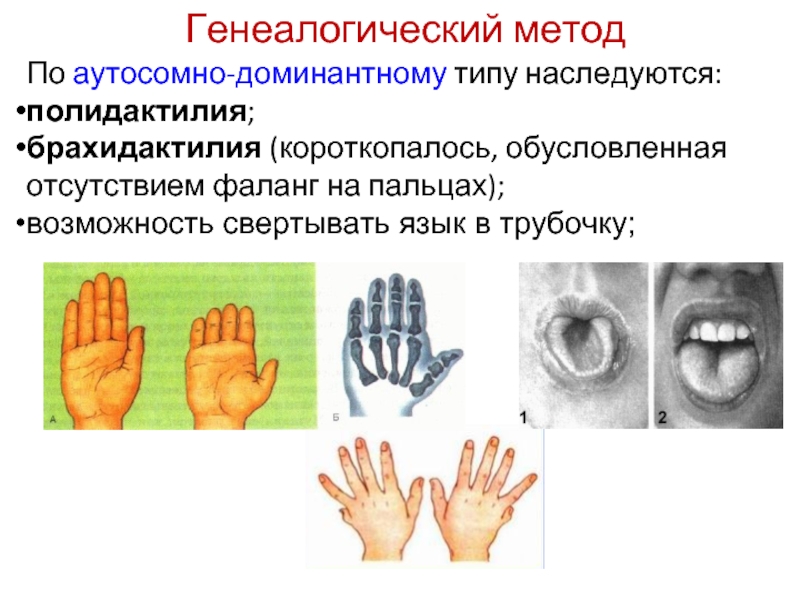



Основная цель операции – это коррекция пальцев на ногах и руках (удлинение укороченных частей, восстановление функций, а также исправление внешних дефектов на кистях и стопах).
Чаще применяются именно микрохирургические операции. В некоторых ситуациях используется специальный аппарат Илизарова. Он помогает удлинить деформированные фаланги, также восстановить нормальную длину нижних и верхних конечностей.
Классификация: виды и типы
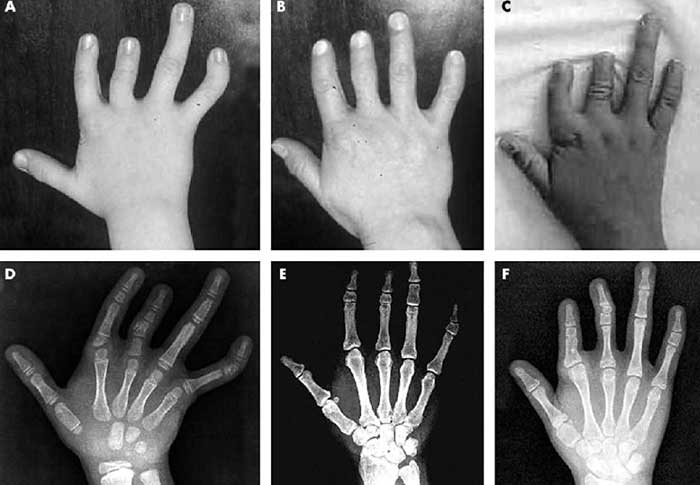
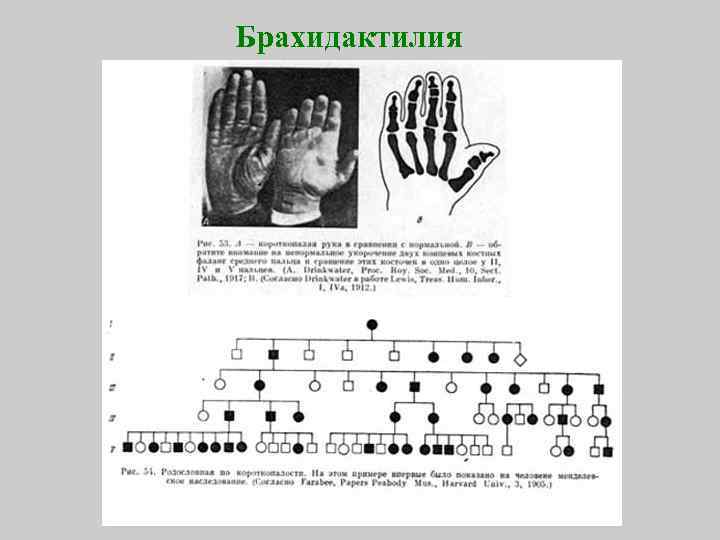
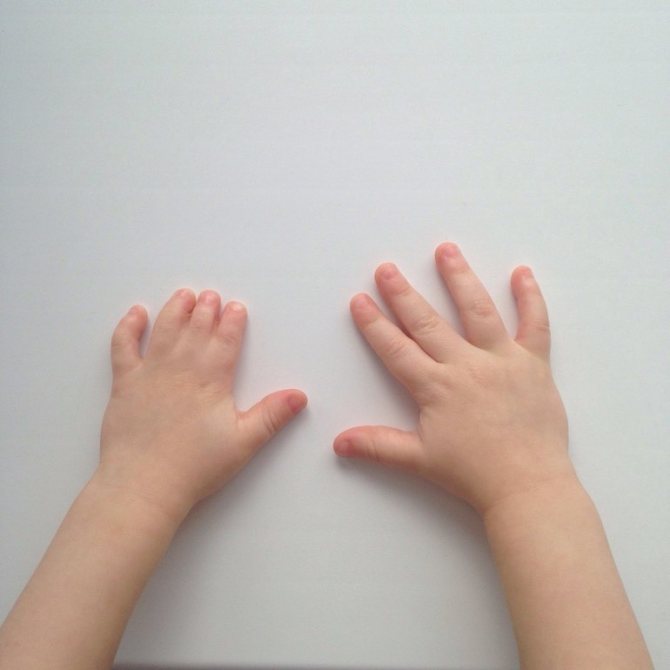
Обычно, короткопалость врач замечают ещё в родильном доме. Но бывают случаи, когда при рождении дефект выражен незначительно, патология списывается на индивидуальную особенность ребёнка. С возрастом брахидактилия становится более выраженной и может причинять малышу как физический, так и психоэмоциональный дискомфорт.
В соответствии с классификацией, различаются определенные разновидности врожденной брахидактилии, которая характеризуется различной степенью поражения фаланг и на различных пальцах. Лишь при определенных вариациях патология способна сочетаться с умственной недоразвитостью малыша и его недостаточным ростом. Зачастую наблюдаются случаи заболевания разновидностей А и D. На умственное развитие они не несут какого-либо вреда.
Типы брахидактилии

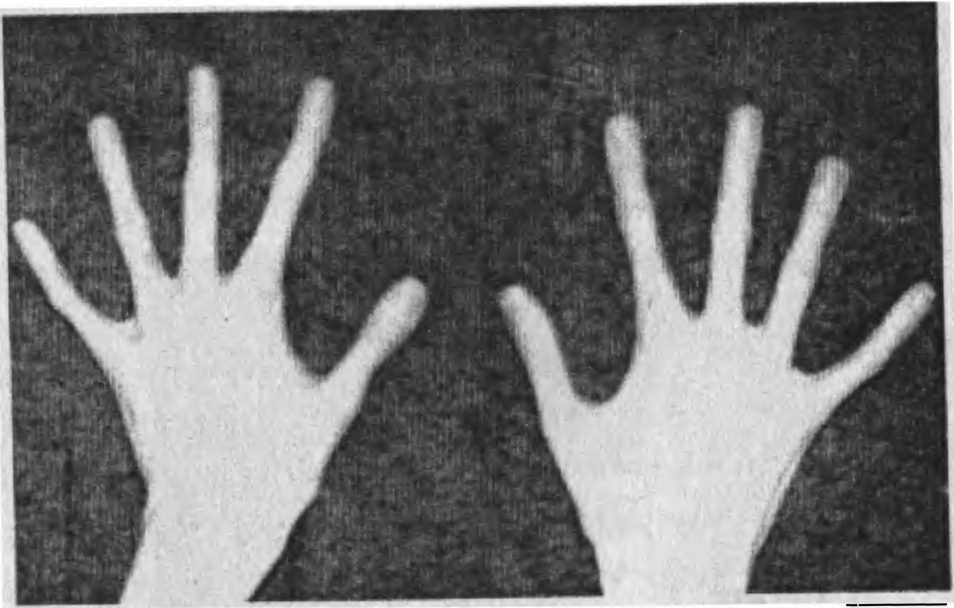
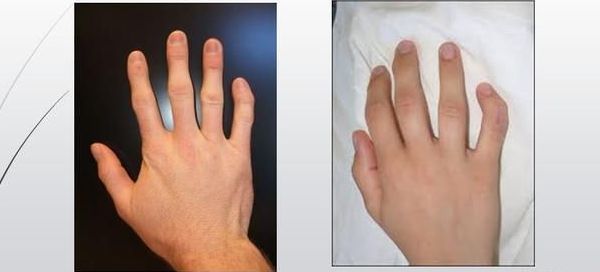
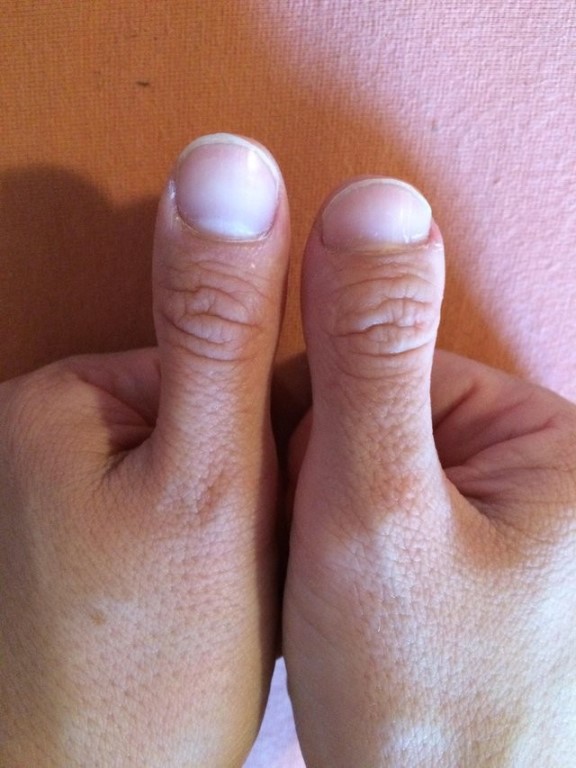
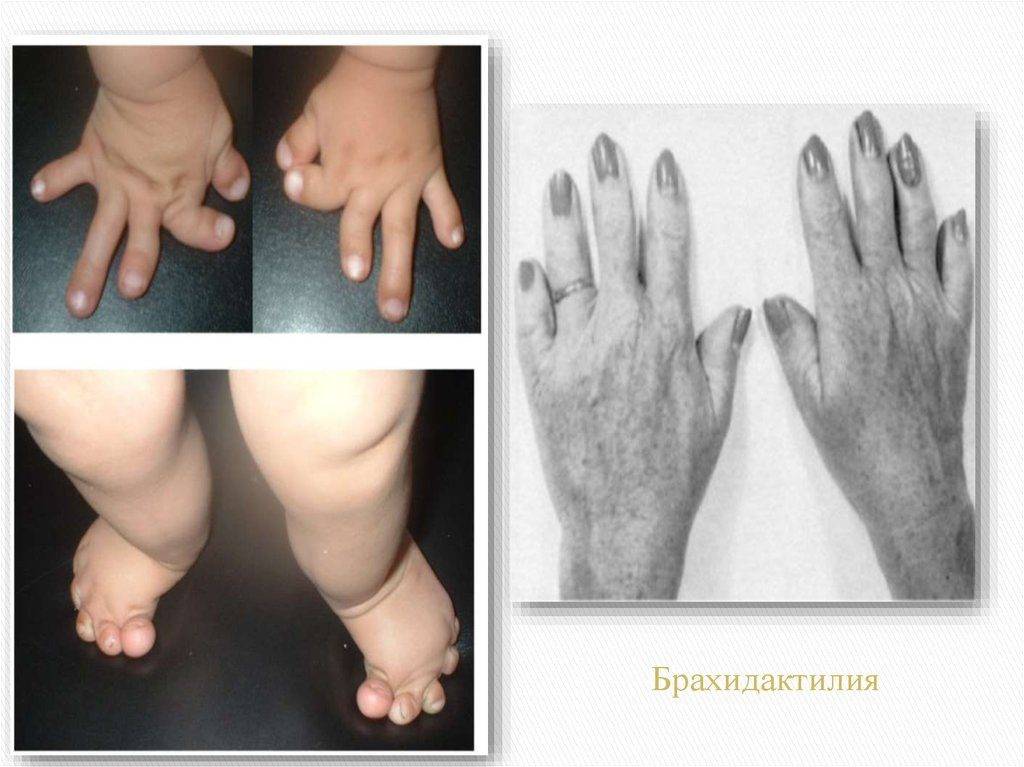
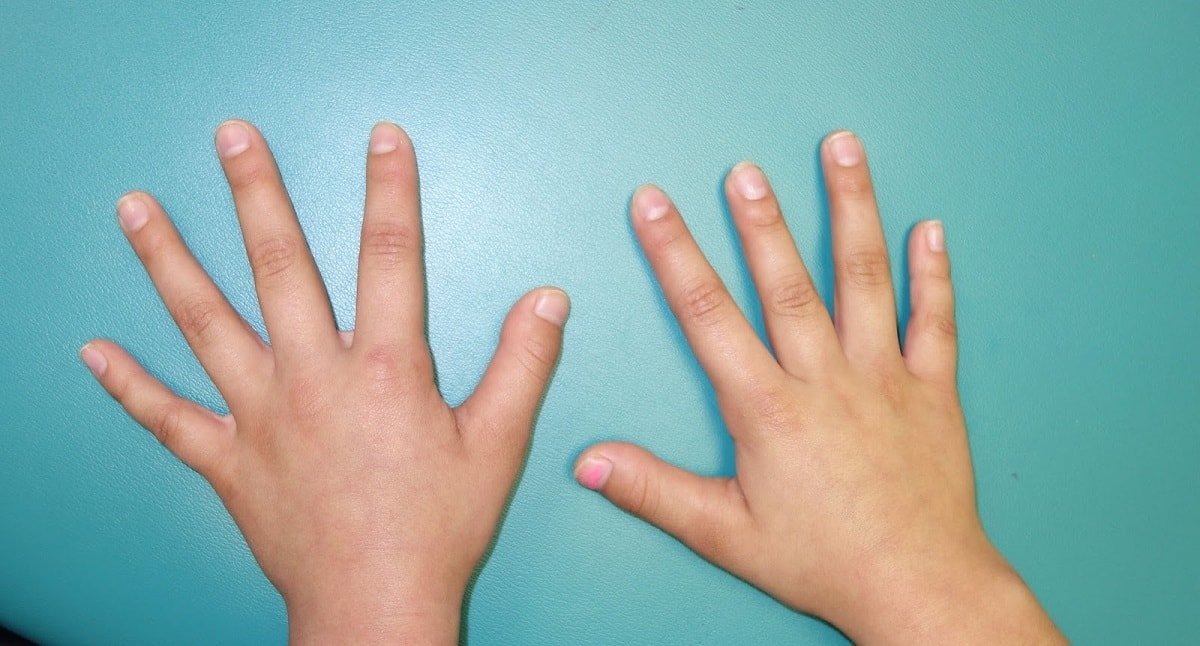
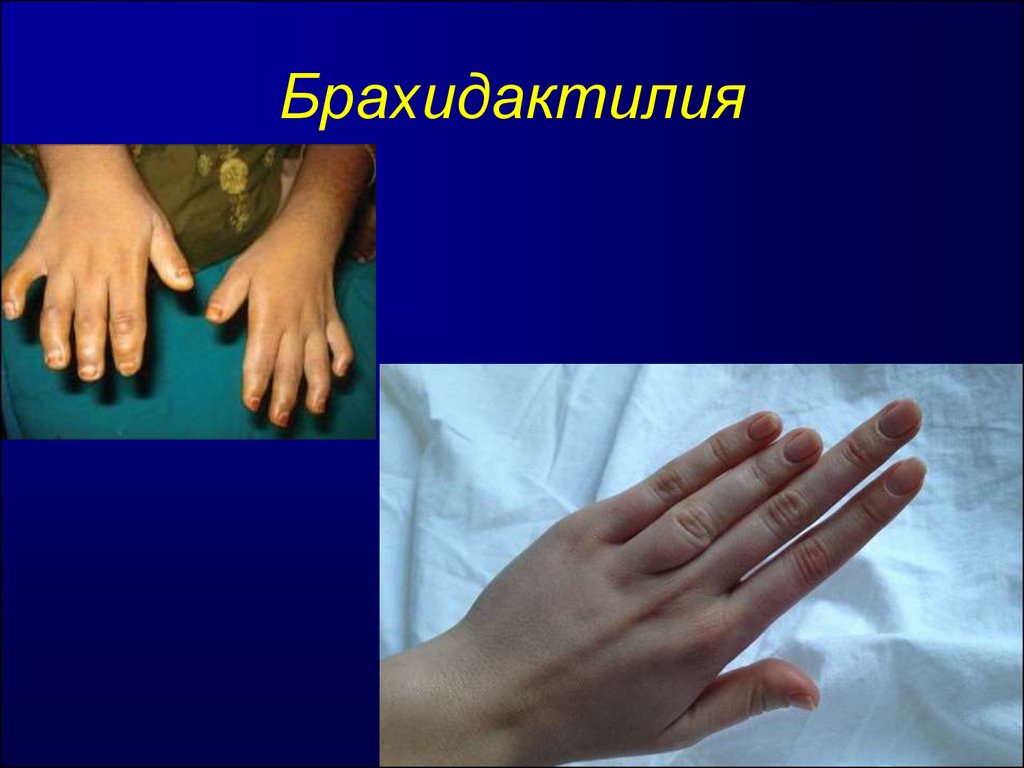
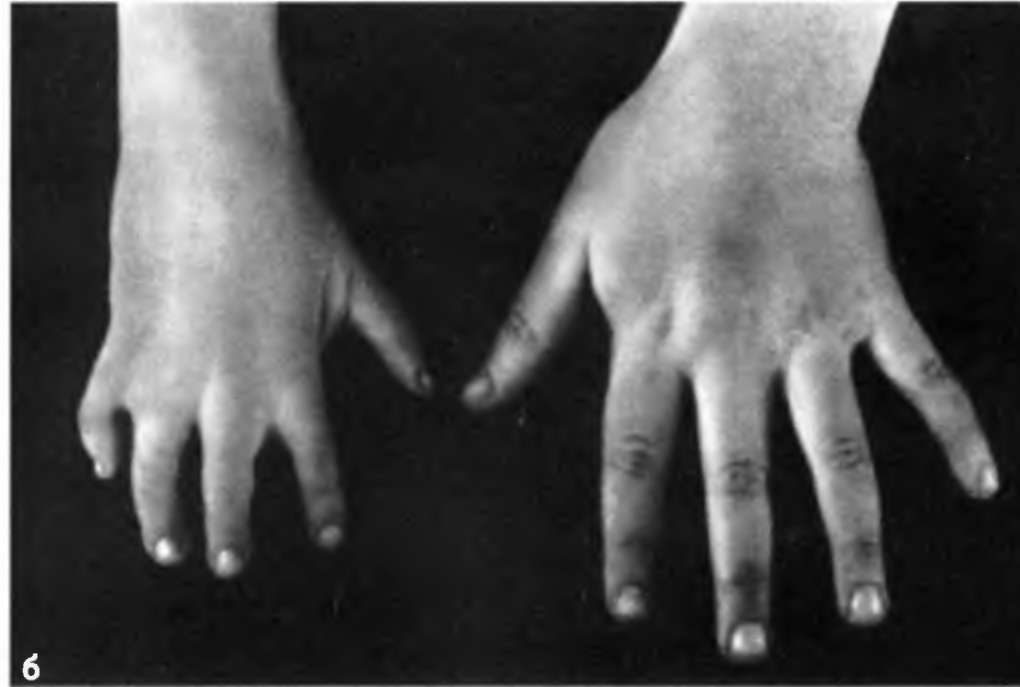
На фото брахидактилия большого пальца Различают несколько клинических вариантов заболевания:
- Тип А. Данный вид брахидактилии отличается поражением средних фаланг, изменением ногтевых пластин и искривлением пальцев. В зависимости от локализации патологии тип А в свою очередь разделяют на 6 подтипов.
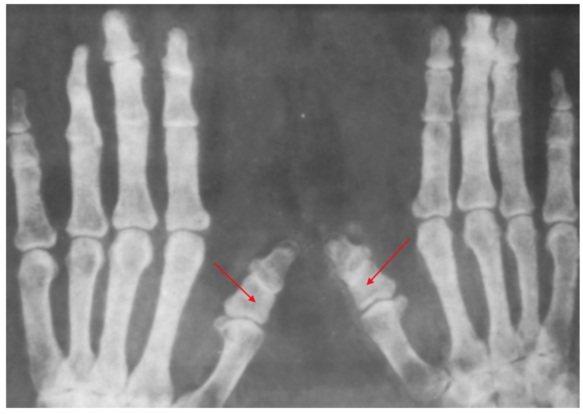
- Тип В. Кроме типичного для типа А поражения средних фаланг пальцев, тип В характеризуется ещё и недоразвитием или отсутствием ногтевых фаланг всех пальцев кистей и стоп. Нередко наблюдается синдактилия – сращение II и III пальцев.
- Тип С – отличительной особенностью является укорочение средних фаланг, а также 2-го и 3-го пальцев с недоразвитостью 1-й пястной кости, что приводит к укорочению большого пальца.
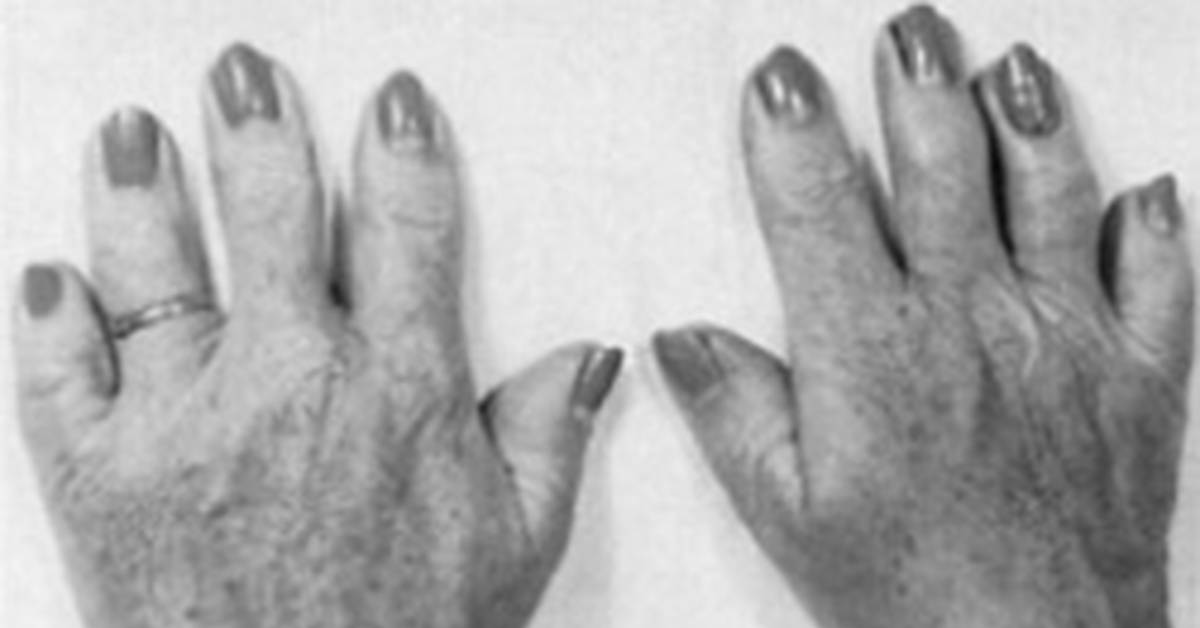
- Тип D – недоразвитость дистальной фаланги, за счет чего происходит укорочение больших пальцев рук и ног. В медицине называется брахимегалодактилия.
- Тип Е – недоразвитость плюсневой и пястной кости.
Брахидактилия А проявляется такими симптомами, как укорочение средних пальцев, дисплазия ногтей и недоразвитие фаланг. Выраженность отклонений в строении может сильно варьироваться, в связи с этим выделяют 5 вариантов короткопалости А:
- А1 – укорочение средних пальцев и предпоследних фаланг больших пальцев рук и ног, задержка роста;
- А 2 – значительное уменьшение фаланг средних пальцев, их ромбовидная форма;
- А 3 – изменение длины и очертаний средних фаланг пятых пальцев верхних конечностей;
- А 4 – недоразвитие костной ткани средних фаланг второго и пятого пальцев рук, косолапость;
- А 5 – отсутствие проксимальных фаланг пятого и второго пальцев, укорочение ногтевых фаланг на руках.
Брахидактилия В отличается деформацией ногтевых фаланг, а также сращиванием второго и третьего пальцев. Поражаются и руки, и ноги. Присутствуют признаки аномального строения позвоночника, зубов и черепа.
Короткопалость С – самый тяжелы тип патологии. Он характеризуется деформацией пястных костей и сращиванием фаланг между собой. Сочетается с низкорослостью и задержкой умственного развития (не всегда).
Брахидактилия большого пальца относится к типу Д. Этот вариант заболевания, затрагивающий руки и ноги, встречается чаще всего.
Короткопалость Е проявляется в виде недоразвития пястных костей и ключиц без изменения фаланг. Наблюдается очень редко.
Симптомы брахидактилии
В ситуации, когда брахидактилия является самостоятельной болезнью, тогда единственным ее симптомом будут укороченные пальцы, что является всего лишь косметическим дефектом, который можно устранить. Однако нередко заболевание является лишь синдромом другой генетической болезни, которая проявляется в своих специфических симптомах.
Достаточно часто брахидактилия сопровождает синдром Дауна. Здесь могут отмечаться такие признаки, как:
- Воронкообразная клетка груди.
- Брахицефалический череп.
- Эпикантус.
- Укорочение шеи.
При синдроме Поланда брахидактилия отмечается деформацией ребер и амастией. При болезни Аарского-Скотта ребенок страдает умственной отсталостью, фимозом, нестабильностью суставов, помимо короткопалости.
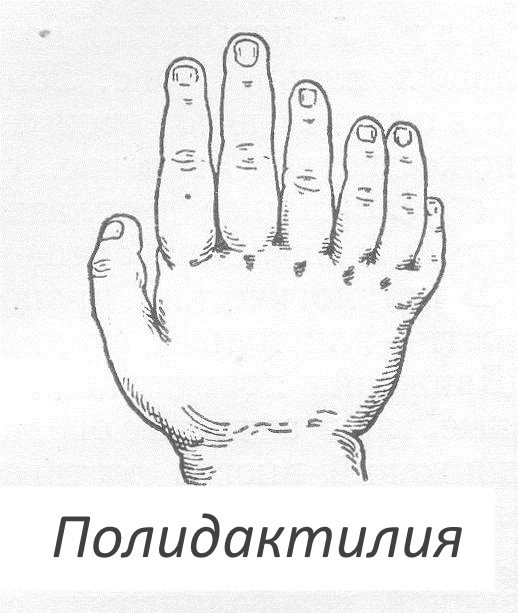
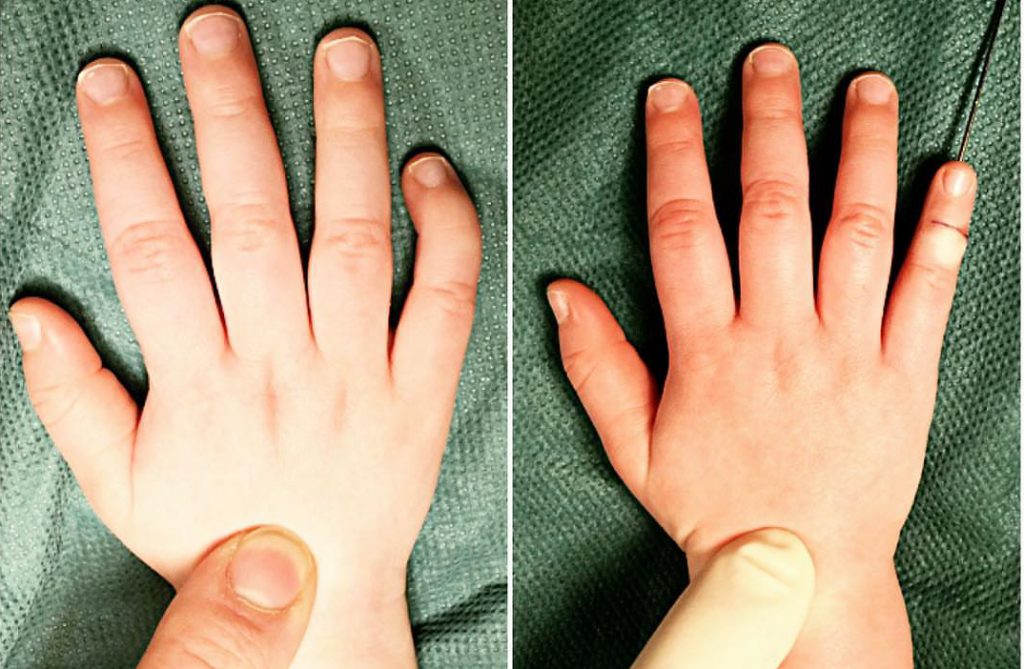
Отсутствие полидактилии и сращения фаланг не ограничивает ребенка в движениях. При серьезных патологиях болезни ограничивается движение межфаланговых суставов. Диспластические повреждения ногтевых пластин также проявляются при болезни, что создает неэстетический вид. Также отмечается необычная конфигурация пальцев в виде расщепления фаланг и уплощения.
перейти наверх
Лечение
Устранить первопричину болезни в настоящее время невозможно, потому для лечения применяют как консервативные, так и хирургические методики, где последние является единственно эффективными.
Они направлены на восстановление двигательной активности и на эстетический результат. В связи с этим устраняются межфаланговые сращения, выполняется костная, сухожильно-мышечная и кожная пластика, увеличиваются линейные размеры стоп и кистей. Применяют следующие оперативные техники: дистракцию, поллинизацию, аутотрансплантацию пальца стопы на кисть. Таким образом, в обязательном порядке потребуется консультация хирурга и ортопеда.
Консервативное лечение направлено на улучшение работы мышечно-связочного аппарата, при этом применяются физиотерапевтические манипуляции, например, лечебная физическая культура, курсы массажа. Их следует рассматривать как дополнительные мероприятия, применяемые в послеоперационном периоде с профилактической целью, чтобы предотвратить вторичную деформацию мелких суставов.
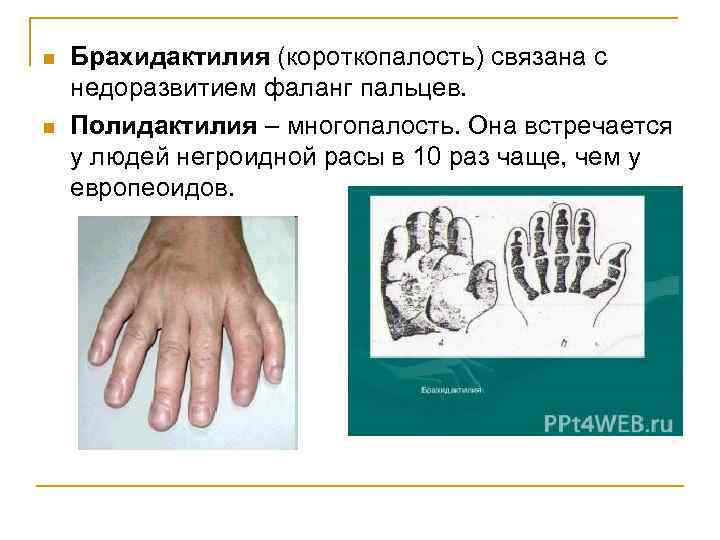

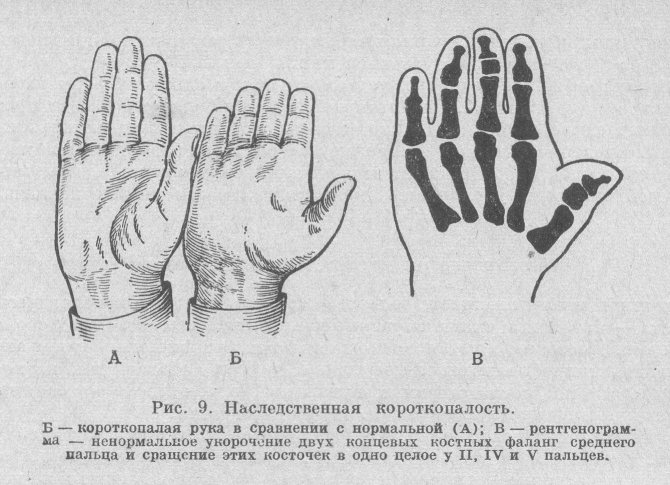

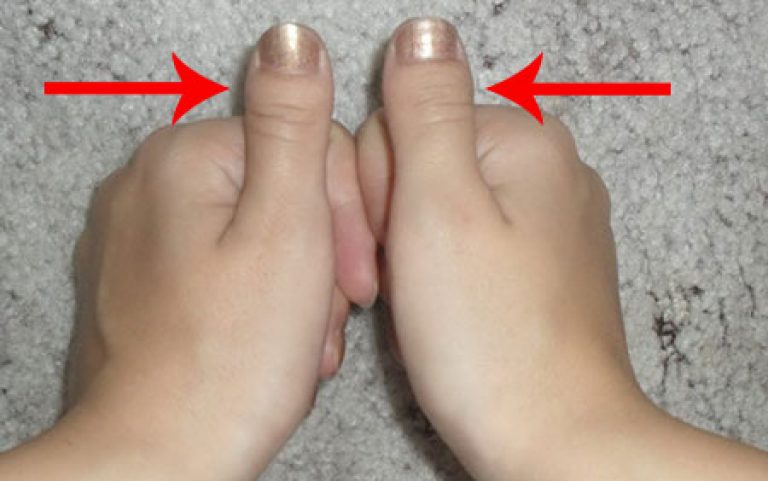
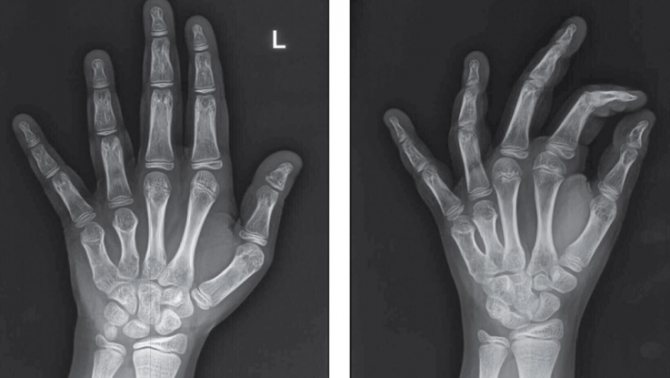
Симптомы
В некоторых случаях брахидактилия является изолированной врожденной аномалией, а иногда короткопалость может сочетаться с другим врожденными синдромами. Так, например, при синдроме Дауна, совместно с брахидактилией, у пациента может выявляться брахицефалия, короткая шея, эпикантус, катаракта, аномалии зубных рядов, бороздчатый язык, врожденные пороки сердца, косоглазие, деформация грудной клетки.
Также, минимальными диагностическими признаками синдрома Беймонда является брахидактилия, нистагм, мозжечковая атаксия, а у больных с синдромом Поланда отсутствует большая и малая грудные мышцы, выявляется брахидактилия, синдактилия, аплазия сосков или амастия, деформация ребер.
Синдром срединной расщелины лица сопровождается выявлением микрогении, атрезии хоан, гипоплазии лобных пазух, брахидактилии, гипофизарного нанизма, вторичного гипотиреоза, двустороннего крипторхизма, искривления полового члена, олигофрении.
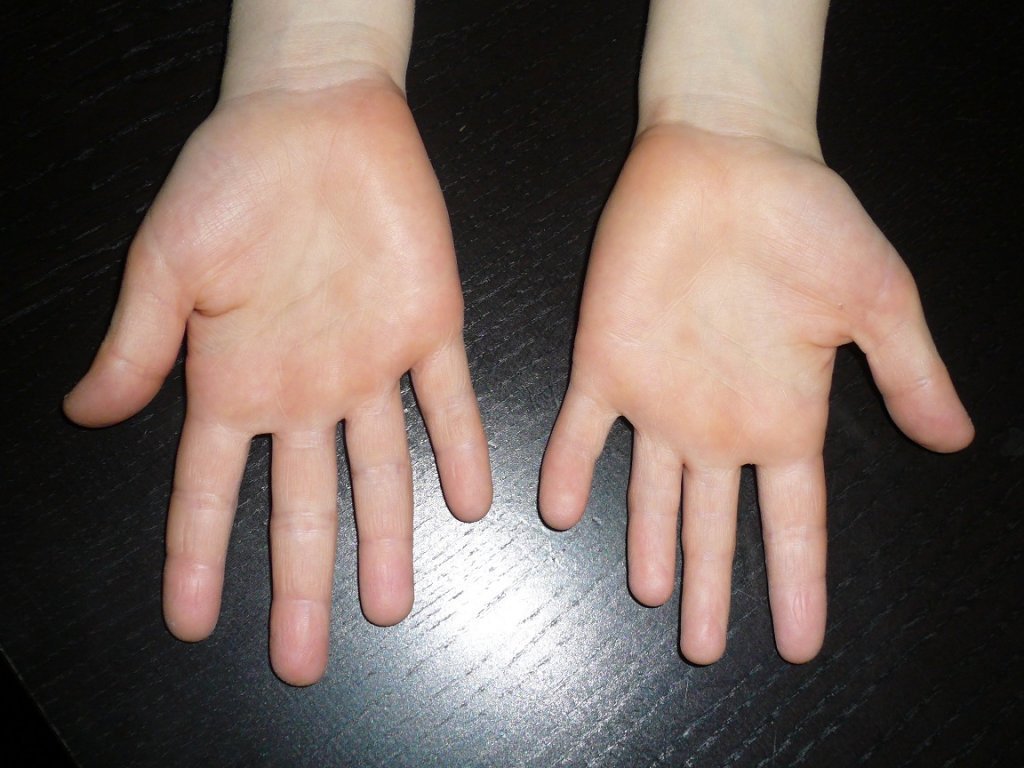
Типичным проявлением брахидактилии является патологическое укорочение пальцев рук и ног, иногда может отмечаться выявление их сплющенной формы или расщепления. В зависимости от тяжести патологии, брахидактилия может сочетаться с тугоподвижностью в суставах, синдактилией, полидактилией, мышечной слабостью, которые могут вызывать нарушение в работе кистей или стоп. При брахидактилии может выявляться гипоплазия или аплазия ногтевых пластин.
Довольно часто брахидактилия сочетается с эктродактилией и адактилией. При эктродактилии наблюдается недоразвитие дистальных фаланг, а в тяжелых случаях может отмечаться отсутствие ногтевых и средних фаланг. Формирование пястных костей не изменено. При адактилии имеет место врожденное отсутствие фаланг пальцев с частичным или полным сохранением пястных костей.
Полногеномный поиск ассоциаций
Полногеномный поиск ассоциаций (GWAS – genome – wide – association study) – исследование ассоциаций между геномными вариантами и различными фенотипическими проявлениями. Анализ ассоциаций генов-кандидатов лежит в основе идентификации генетических вариантов предрасположенности.
Известно около 90 наследуемых заболеваний и синдромов, сопровождающихся алопецией или гипотрихозом. В большинстве случаев врожденный гипотрихоз является одним из признаков эктодермальных дисплазий, изолированных или сочетанных. В тех случаях, когда изменения волос доминируют и имеют большое диагностическое значение, их рассматривают как отдельное заболевание.
Затем Сафонова Л.А. подробнее рассмотрела некоторые заболевания волосистой части головы.
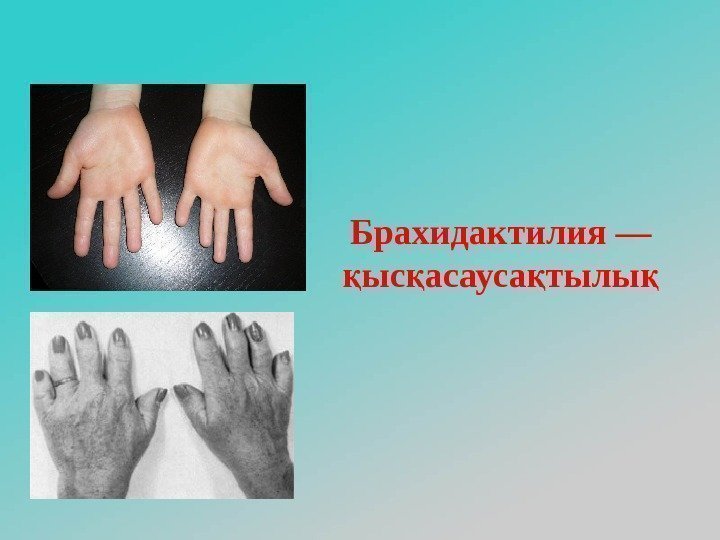
Описание и причины болезни
Данная патология проявляется в виде деформации костей кисти и стопы. Недоразвитые фаланги внешне выглядят укороченными. Причины возникновения такого дефекта не очень разнообразны. Обычно это передача мутантного гена малышу от одного из его родителей. Она в одинаковых долях припадает как на представителей мужского, так и женского пола.
Процент таких патологий среди всех наследственных недугов достаточно высок — практически четвертая часть. Если другие аномалии у человека отсутствуют, он ничем не отличается от остальных людей, может жить нормальной жизнью. Но чаще всего болезнь возникает вместе с другими отклонениями:
- Синдром Дауна. Наблюдается укорочение шеи и черепа, аномалии челюсти, катаракта и косоглазость, проблемы с сердцем и дефекты грудной клетки.
- Синдром Поланда. Происходит деформация ребер и мышц груди, полное отсутствие молочных желез.
- Синдром Беймонда. Присутствие мозжечковой атаксии, частого непроизвольного движения глаз.
- Синдром Аарскога-Скотта. Отмечается неустойчивость суставов, задержка либо неполное развитие психики, грыжи в области паха, сращивание пальцев.
При наличии одного из таких заболеваний, кроме визуального дефекта, к недугу добавляются и более серьезные симптомы. С помощью ранней диагностики можно выявить отклонение у плода еще при беременности.
Значение исследования CAG повторов гена AR в общей практике
Исследования полиморфизма CAG-повторов в 1 экзоне гена андрогенетического рецептора (AR) имеет диагностическое и прогностическое значение, позволяющее определить риск репродуктивных нарушений, рака простаты, установить причину мужского бесплодия, мышечной атрофии.
Диапазон 20 — 26 повторов считается относительной нормой. При уменьшенном количестве CAG-повторов (<18) в гене AR отмечается повышенная чувствительность к андрогенам и увеличивается риск рака простаты. А у женщин в этом случае отмечается повышенный риск развития рака молочной железы, причем более агрессивной формы, рака эндометрия, СПКЯ и гиперандрогении.
Увеличение количества CAG-повторов (>26) свидетельствует о наличии генетической предрасположенности к гормонозависимому нарушению сперматогенеза.
Увеличение числа CAG-повторов до 38 — 62 приводит к спинобульбарной мышечной атрофии типа Кеннеди.
Результаты генетического теста позволяют врача внедрять предсимптоматическую оценку риска, что позволяет провести раннюю диагностику и предотвратить наступление заболевания, предложив персонализированные рекомендации. Синтез традиционных методов обследования и результатов генетического тестирования позволяет персонифицировать рекомендации по применению лечебных мероприятий и их длительность.
Синдром Менкеса
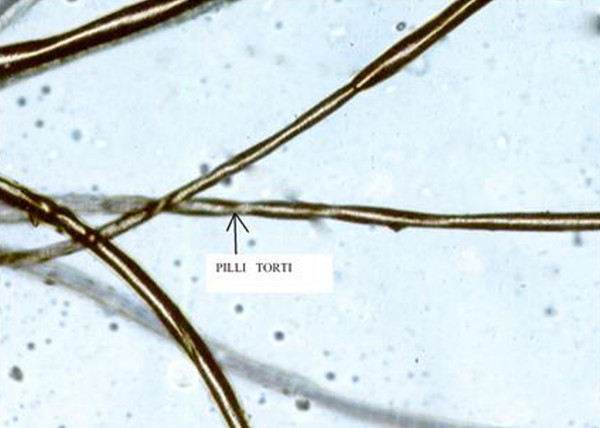 | 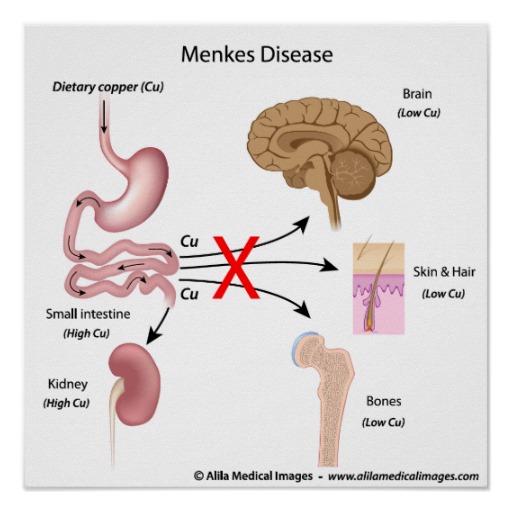 |
Развитие синдрома связано с дефицитом меди вследствие нарушения ее абсорбции. Заболевание наследуется рецессивно, сцеплено с Х-хромосомой. Нарушается работа АТР фермента, обеспечивающего доставки CU2+. Значительное снижение содержание меди отмечается в сыворотке крови, ткани мозга и печени. В плазме резко снижено содержание церулоплазмина. В то же время высокие концентрации Сu2+ обнаруживают в тканях пищевода, почек, легких, селезенки, поджелудочной железы, коже и мышцах. В основном болеют мальчики.
Клинические проявления заболевания:
структурные аномалии стержня волос, гипотрихоз
тяжелая умственная и физическая отсталость
задержка роста и патология костной ткани
судороги
высокая подверженность инфекциям
аномалии почек и сосудов, зрительные расстройства
диагноз ставят на основании клинической картины, данных световой микроскопии волос, биохимического анализа крови
Важно своевременное распознавание болезни, так как парентеральное введения препарата Cu2+ дает обнадеживающие результаты только при очень раннем их назначении — в течение первого месяца жизни. При уже возникших изменениях в ЦНС клинического эффекта не наблюдается, даже при нормализации уровня Cu2+ в плазме крови
При уже возникших изменениях в ЦНС клинического эффекта не наблюдается, даже при нормализации уровня Cu2+ в плазме крови.
Лечение брахидактилии

В лечении короткопалости применяются консервативные и хирургические методы. Нередко с целью восстановления функционирования кисти или стопы достаточно проведения курсов лечебной физкультуры и массажа. Это способствует улучшению работы мышечно-связочного аппарата и позволяет избежать значительных ограничений двигательной активности.
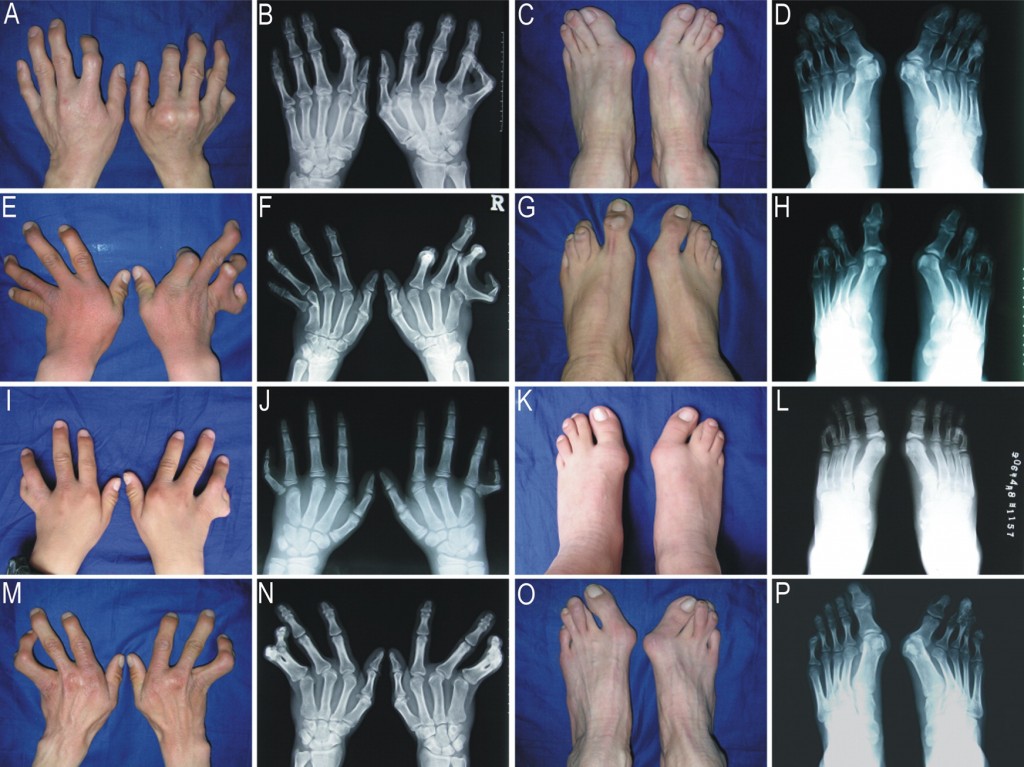
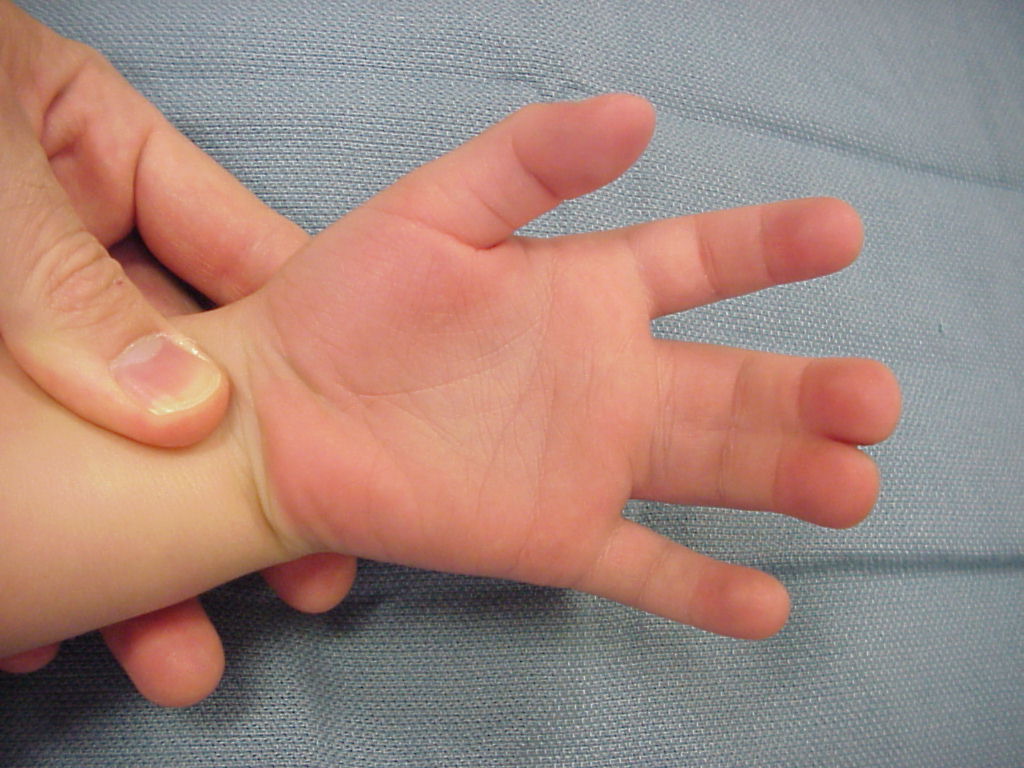
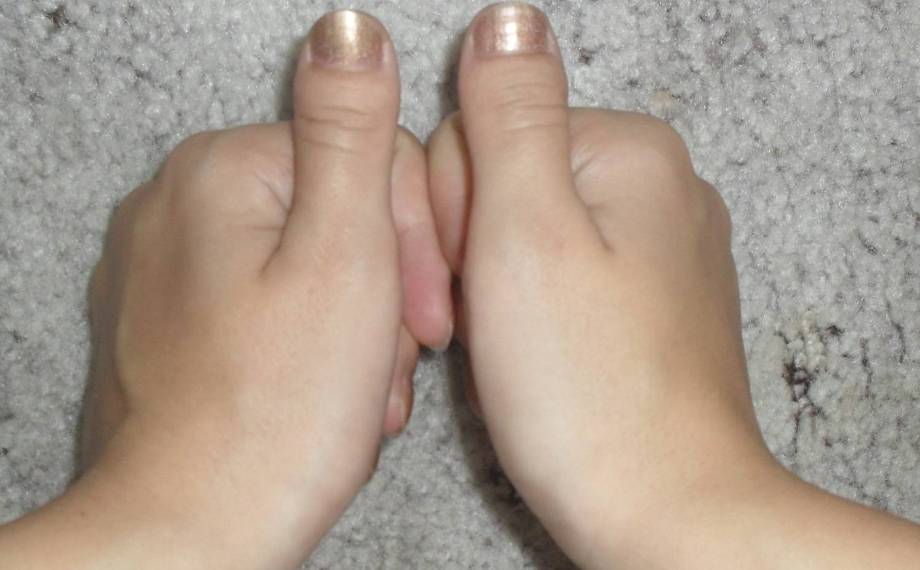
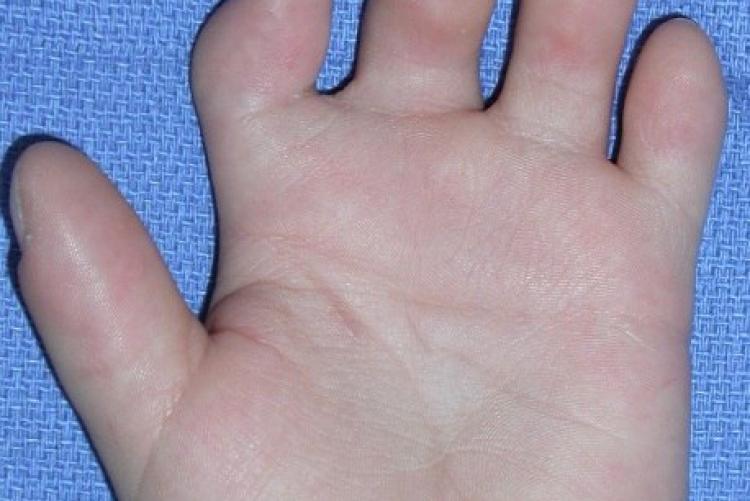
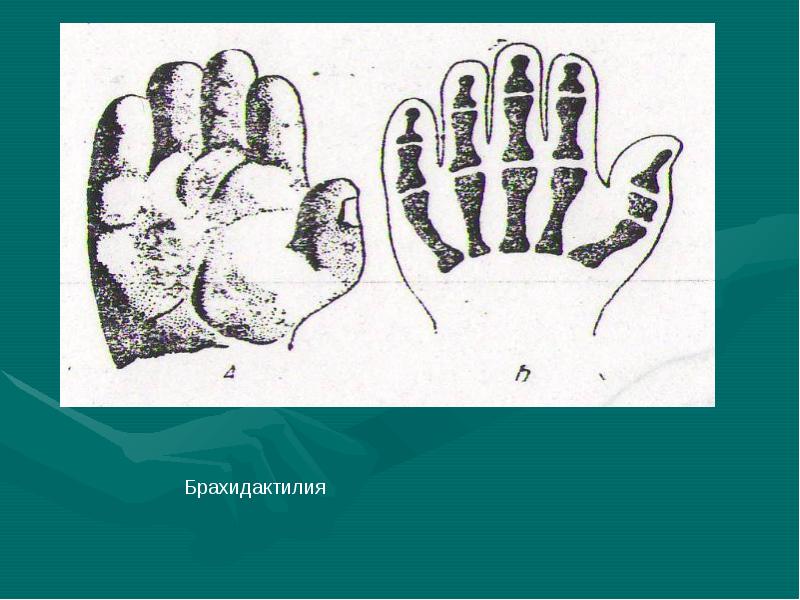
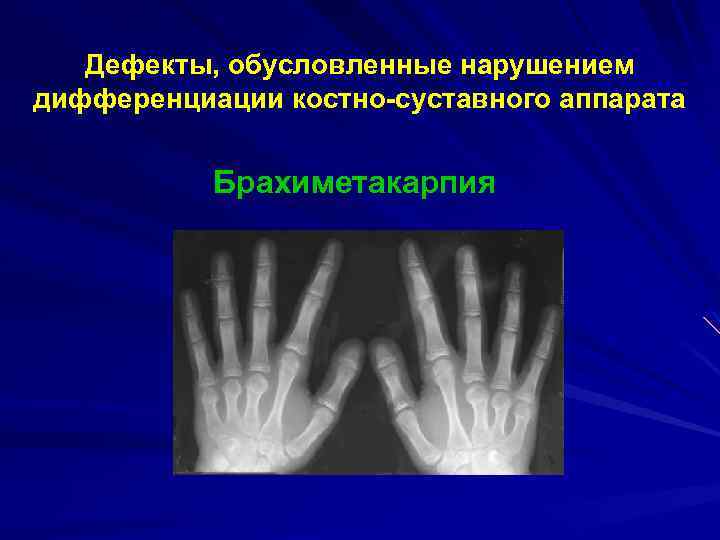
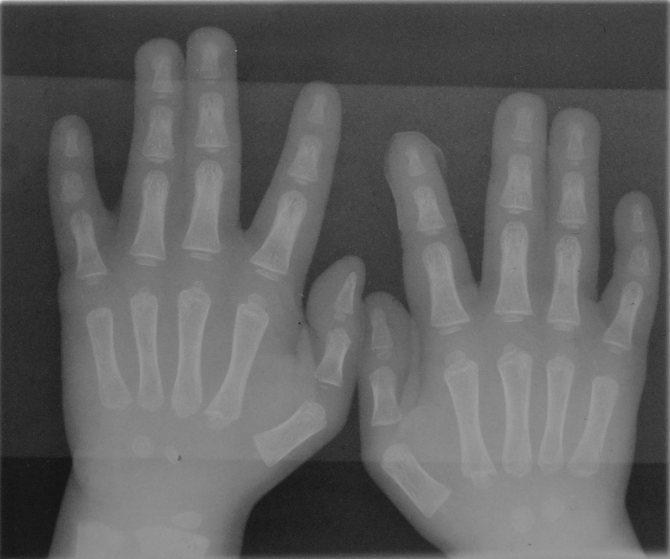

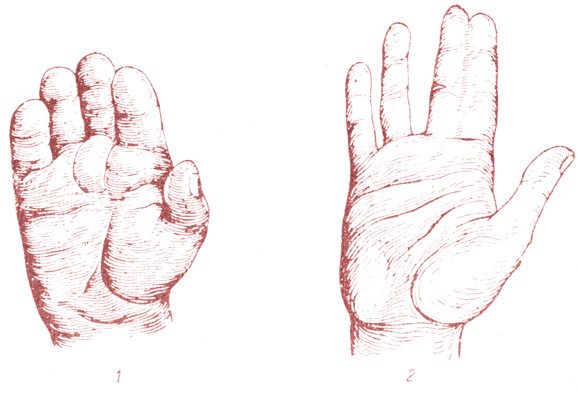

При хирургической коррекции брахидактилии основной целью служит изменение размеров кисти или стопы, а также устранение сопутствующей аномалии развития. Например, при синдактилии операция подразумевает разделение сросшихся пальцев с дальнейшей кожной и сухожильно-мышечной пластикой. Хирургическое лечение заболевания, прежде всего, направлено на максимально возможное восстановление функций и уже вторично — на достижение эстетического результата.
Брахидактилия – это наследственная патология, поэтому важно помнить, что с целью её предотвращения необходимо исключение родственных браков
Диагностика
Заболевание возможно выявить ещё во время вынашивания ребёнка, подтверждение диагноза происходит после появления малыша на свет. Поэтому выявление патологии складывается из нескольких этапов:
генетическое консультирование.
Семьям, в которых встречались случаи рождения ребёнка с патологией костной системы обязательно нужно пройти консультацию у врача-генетика во время планирования беременности. Врач поможет рассчитать риск рождения ребёнка с данной аномалией, порекомендует исследовать генотип родителей, чтобы удостоверится в отсутствии мутаций;
обследование беременной женщины.
С помощью УЗИ врач может обнаружить и оценить патологию ещё во время вынашивания ребёнка. Кроме того во время исследования оценивается состояние и функция других органов и систем, развитие малыша.
Изолированная брахидактилия не считается тяжёлым пороком и не требует прерывания беременности, но служит поводом заподозрить наличие у ребёнка наследственного синдрома. Многие из них (синдром Дауна, Поланда, Беймонда и другие) помимо основных проявлений, пороков развития сопровождаются укорочением пальцев кистей и стоп.
Чтобы заподозрить опасное заболевание у ребёнка результаты УЗИ оцениваются вместе с биохимическим скринингом. При необходимости проводятся инвазивные методы исследования, способные с точностью до 99% установить наличие наследственного синдрома у малыша;
обследование малыша после рождения.
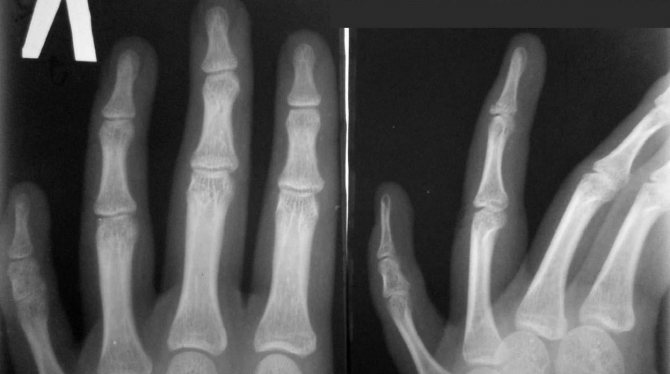
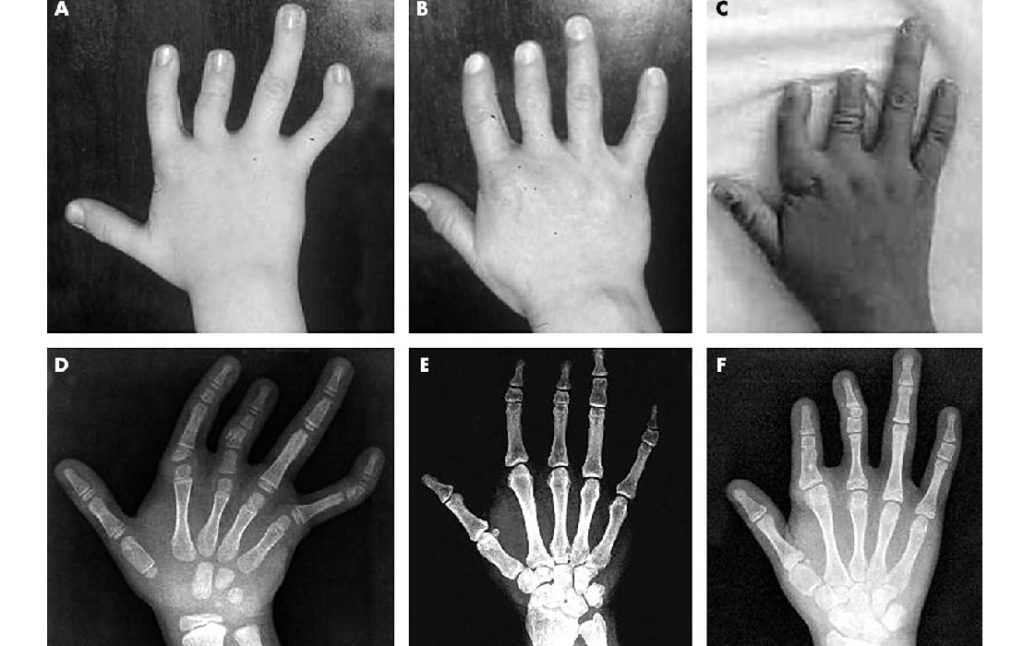
Обследование ребёнка складывается из сбора анамнеза заболевания, тщательного осмотра костно-суставной системы малыша, инструментальных методов диагностики. Самым простым и надёжным способом определить патологию остаётся рентгенологическое исследование. При необходимости врач может порекомендовать провести компьютерную томографию.
В случаях сомнительного диагноза, для исключения наследственного синдрома у крохи, проводится кариотипирование. С помощью данного метода определяются нарушения в геноме малыша, строении и количестве хромосом.